Содержание книги "Ультразвуковая топографическая анатомия периферической нервной системы"
1 Как извлечь пользу из книги: «Руководство пользователя»!
2 Шея
3 Плечо, предплечье и кисть
4 Туловище
5 Ягодичная область
6 Бедро, голень и стопа
Книга содержит всего пять основных разделов:
- Шея
- Верхняя конечность (плечо, предплечье/кисть)
- Туловище
- Ягодичная область
- Нижняя конечность (бедро, голень/стопа)
Нервы, проходящие в этих анатомических областях, перечисляются в алфавитном порядке (с номерами страниц) на латыни в соответствии с Terminología Anatómica (Международной анатомической терминологией) FCAT (Федеративного международного комитета по анатомической терминологии; 1998 IFFA; FACT; издательство Georg Thieme, Штутгарт, ФРГ) и описываются в таком порядке, каждый нерв на отдельном раз-вороте книги:
- Основные ветви (ramus/rami) необходимо искать по наименованию нерва, дающего им начало!
- Нерву, упоминаемому повторно, присваивается порядковый номер, а также приводится наименование анатомической области!
Пример: ладонная ветвь срединного нерва (r. palmaris n. mediani)
Пошаговый алгоритм поиска соответствующей ветви:
1. Выберите анатомическую область - см. раздел «Предплечье/кисть».
2. Найдите в списке заголовок «Nervus medianus (срединный нерв)».
3. Найдите подзаголовок «Nervus medianus, r. palmaris (ладонная ветвь срединного нерва)» с указанными номерами страниц.
4. Для ознакомления с материалом откройте указанную страницу. и. Руководство пользователя!
Вступление к книге "Ультразвуковая топографическая анатомия периферической нервной системы"
Если мы хотим научиться безошибочно находить и идентифицировать периферические нервы при проведении местной анестезии, лечении болевого синдрома и выполнении других клинических манипуляций, нам необходимы глубокие познания в области клинической анатомии. Ориентироваться при поиске нервов помогают находящиеся рядом с ними крупные сосуды, мышцы, костные образования и другие структуры. Для начинающих специалистов в области эхографии, изучающих ультразвуковую диагностику и анатомию нервной системы, непросто самостоятельно и быстро находить нужные анатомические образования. Этот процесс часто напоминает навигацию по незнакомой местности без карты и компаса. Такое путешествие требует тщательной подготовки, сбора информации и ознакомления с ландшафтом. Это особенно верно для проведения регионарной анестезии под ультразвуковым контролем и прочих интервенционных методов обезболивания или диагностики. Данный иллюстрированный справочник по ультразвуковому исследованию (УЗИ) периферической нервной системы составлен профессорами Hannes Gruber, Alexander Loizides, Bernhard Moriggl и другими экспертами в области УЗИ периферических нервов и служит этаким спасительным путеводителем по неизведанному или своеобразными картами «Google» для ультразвуковой навигации по периферической нервной системе. Справочник написан доступным языком, имеет простую структуру и содержит иллюстрации с основными анатомическими ориентирами, а также практические рекомендации по технике исследования и стандартные алгоритмы поиска нервов. Книга освещает основные и альтернативные алгоритмы поиска нервов с учетом вариабельности анатомии человека, на которую часто ссылается профессор Moriggl, лучший из известных мне специалистов в области ультразвуковой анатомии. Подобный ультразвуковой атлас - отличный гид по малоизученным анатомическим ландшафтам. Я уверен, что эта уникальная книга - бесценный помощник в изучении ультразвуковой диагностики, анатомии опорно-двигательного аппарата, а также периферической нервной системы. К вам на помощь придут глубочайшие познания авторов и их практические советы по технике исследования с пошаговыми алгоритмами поиска отдельных нервов. Справочник, отличающийся лаконичностью и глубиной подачи материала, подготовит вас к путешествию по неизведанным территориям периферической нервной системы. И пусть это издание станет вашей настольной книгой! Vincent W.S. Chan, MD, FRCPC, FRCA Отделение анестезиологии Университет Торонто Торонто, Онтарио, Канада
«Для кого и зачем?» Целевая аудитория и предназначение этой книги. Эта книга задумана как карманный путеводитель для коллег, уже практикующих УЗИ нервов и еще только интересующихся этой методикой. Она позволит быстро найти любые нервы в повседневной клинической практике. Можно выразиться и так: тебе больше не нужно искать нерв, ибо ты его уже нашел в этой книге. Таким образом, ответ на вопрос «зачем?» уже почти готов, ведь подобные руководства еще не издавались. Наша книга позволит сэкономить драгоценное время для диагностики, инвазивных вмешательств и/или планового лечения. Четкие и краткие (!) иллюстрированные описания видимых и/или пальпируемых «внешних» ориентиров позволят быстро установить ультразвуковой датчик в оптимальной исходной позиции. На УЗ-изображениях отмечены лишь основные «внутренние» ориентиры, позволяющие локализовать нерв и охарактеризовать его топографическое местоположение среди других структур. В практических целях мы выделили зоны, где наиболее четко визуализируются отдельные нервы и ветви, назвав их «Точками Оптимальной Видимости» (ТОВ). Практически каждый (!) периферический нерв имеет свою ТОВ, которая приводится в обзорных таблицах по отдельным нервам. Твердое знание ТОВ поможет ориентироваться в сложных ситуациях, особенно при плохом акустическом доступе. Авторы уделили особенное внимание практическим моментам УЗИ периферических нервов, что позволило им описать анатомические варианты прохождения нервов и постараться рассмотреть альтернативные алгоритмы их поиска. Дополняют книгу комментарии по позициям датчика и частым техническим ошибкам. Ссылки на бесчисленные исследования по УЗИ периферических нервов опущены намеренно, так как эти сведения воспрепятствовали бы краткости изложения материала. Мы очень надеемся, что этот справочник придется вам по душе и станет вашей настольной книгой! А завершим мы наше вступительное слово вариацией на тему цитаты известного немецкого писателя Вильгельма Буша (1832-1908): Вы никогда не найдете нерв там, где его никогда не было. Пусть наша книга поможет вам избежать этой ошибки! Инсбрук, Австрия Bernhard Moriggl Alexander Loizides Hannes Gruber
Nervus ulnaris: г. dorsalis manus (локтевой нерв: тыльная ветвь кисти)
Рис. 3.89 Отведение кисти в локтевую сторону облегчает пальпацию локтевого сгибателя запястья (m. flexor carpi ulnaris)
Рис. 3.90 Датчик устанавливают поперек предплечья ближе к локтевой поверхности, на границе его средней и дистальной третей
Рис. 3.91 Локтевой сгибатель запястья (m. flexor carpi ulnaris; MFCU) с его сухожилием (T-MFCU) идут поверх тыльной ветви сразу после ее отхождения от локтевого нерва. AU: локтевая артерия (a. ulnaris)
Рис. 3.92 ТОВ тыльной ветви локтевого нерва (г. dorsalis manus n. ulnaris; большая стрелка) располагается сразу после места ее отхождения от локтевого нерва (n. ulnaris; маленькие стрелки)
Содержание книги "Ультразвуковая топографическая анатомия периферической нервной системы"
1 Как извлечь пользу из книги: «Руководство пользователя»!
2 Шея
3 Плечо, предплечье и кисть
4 Туловище
5 Ягодичная область
6 Бедро, голень и стопа
Книга содержит всего пять основных разделов:
- Шея
- Верхняя конечность (плечо, предплечье/кисть)
- Туловище
- Ягодичная область
- Нижняя конечность (бедро, голень/стопа)
Нервы, проходящие в этих анатомических областях, перечисляются в алфавитном порядке (с номерами страниц) на латыни в соответствии с Terminología Anatómica (Международной анатомической терминологией) FCAT (Федеративного международного комитета по анатомической терминологии; 1998 IFFA; FACT; издательство Georg Thieme, Штутгарт, ФРГ) и описываются в таком порядке, каждый нерв на отдельном раз-вороте книги:
- Основные ветви (ramus/rami) необходимо искать по наименованию нерва, дающего им начало!
- Нерву, упоминаемому повторно, присваивается порядковый номер, а также приводится наименование анатомической области!
Пример: ладонная ветвь срединного нерва (r. palmaris n. mediani)
Пошаговый алгоритм поиска соответствующей ветви:
1. Выберите анатомическую область - см. раздел «Предплечье/кисть».
2. Найдите в списке заголовок «Nervus medianus (срединный нерв)».
3. Найдите подзаголовок «Nervus medianus, r. palmaris (ладонная ветвь срединного нерва)» с указанными номерами страниц.
4. Для ознакомления с материалом откройте указанную страницу. и. Руководство пользователя!
Вступление к книге "Ультразвуковая топографическая анатомия периферической нервной системы"
Если мы хотим научиться безошибочно находить и идентифицировать периферические нервы при проведении местной анестезии, лечении болевого синдрома и выполнении других клинических манипуляций, нам необходимы глубокие познания в области клинической анатомии. Ориентироваться при поиске нервов помогают находящиеся рядом с ними крупные сосуды, мышцы, костные образования и другие структуры. Для начинающих специалистов в области эхографии, изучающих ультразвуковую диагностику и анатомию нервной системы, непросто самостоятельно и быстро находить нужные анатомические образования. Этот процесс часто напоминает навигацию по незнакомой местности без карты и компаса. Такое путешествие требует тщательной подготовки, сбора информации и ознакомления с ландшафтом. Это особенно верно для проведения регионарной анестезии под ультразвуковым контролем и прочих интервенционных методов обезболивания или диагностики. Данный иллюстрированный справочник по ультразвуковому исследованию (УЗИ) периферической нервной системы составлен профессорами Hannes Gruber, Alexander Loizides, Bernhard Moriggl и другими экспертами в области УЗИ периферических нервов и служит этаким спасительным путеводителем по неизведанному или своеобразными картами «Google» для ультразвуковой навигации по периферической нервной системе. Справочник написан доступным языком, имеет простую структуру и содержит иллюстрации с основными анатомическими ориентирами, а также практические рекомендации по технике исследования и стандартные алгоритмы поиска нервов. Книга освещает основные и альтернативные алгоритмы поиска нервов с учетом вариабельности анатомии человека, на которую часто ссылается профессор Moriggl, лучший из известных мне специалистов в области ультразвуковой анатомии. Подобный ультразвуковой атлас - отличный гид по малоизученным анатомическим ландшафтам. Я уверен, что эта уникальная книга - бесценный помощник в изучении ультразвуковой диагностики, анатомии опорно-двигательного аппарата, а также периферической нервной системы. К вам на помощь придут глубочайшие познания авторов и их практические советы по технике исследования с пошаговыми алгоритмами поиска отдельных нервов. Справочник, отличающийся лаконичностью и глубиной подачи материала, подготовит вас к путешествию по неизведанным территориям периферической нервной системы. И пусть это издание станет вашей настольной книгой! Vincent W.S. Chan, MD, FRCPC, FRCA Отделение анестезиологии Университет Торонто Торонто, Онтарио, Канада
«Для кого и зачем?» Целевая аудитория и предназначение этой книги. Эта книга задумана как карманный путеводитель для коллег, уже практикующих УЗИ нервов и еще только интересующихся этой методикой. Она позволит быстро найти любые нервы в повседневной клинической практике. Можно выразиться и так: тебе больше не нужно искать нерв, ибо ты его уже нашел в этой книге. Таким образом, ответ на вопрос «зачем?» уже почти готов, ведь подобные руководства еще не издавались. Наша книга позволит сэкономить драгоценное время для диагностики, инвазивных вмешательств и/или планового лечения. Четкие и краткие (!) иллюстрированные описания видимых и/или пальпируемых «внешних» ориентиров позволят быстро установить ультразвуковой датчик в оптимальной исходной позиции. На УЗ-изображениях отмечены лишь основные «внутренние» ориентиры, позволяющие локализовать нерв и охарактеризовать его топографическое местоположение среди других структур. В практических целях мы выделили зоны, где наиболее четко визуализируются отдельные нервы и ветви, назвав их «Точками Оптимальной Видимости» (ТОВ). Практически каждый (!) периферический нерв имеет свою ТОВ, которая приводится в обзорных таблицах по отдельным нервам. Твердое знание ТОВ поможет ориентироваться в сложных ситуациях, особенно при плохом акустическом доступе. Авторы уделили особенное внимание практическим моментам УЗИ периферических нервов, что позволило им описать анатомические варианты прохождения нервов и постараться рассмотреть альтернативные алгоритмы их поиска. Дополняют книгу комментарии по позициям датчика и частым техническим ошибкам. Ссылки на бесчисленные исследования по УЗИ периферических нервов опущены намеренно, так как эти сведения воспрепятствовали бы краткости изложения материала. Мы очень надеемся, что этот справочник придется вам по душе и станет вашей настольной книгой! А завершим мы наше вступительное слово вариацией на тему цитаты известного немецкого писателя Вильгельма Буша (1832-1908): Вы никогда не найдете нерв там, где его никогда не было. Пусть наша книга поможет вам избежать этой ошибки! Инсбрук, Австрия Bernhard Moriggl Alexander Loizides Hannes Gruber
Ganglion cervicale medium (средний шейный узел симпатического ствола)
Рис. 2.1 Найдите точку пульсации общей сонной артерии у переднего края грудино-ключично-сосцевидной мышцы (m. stemo-cleidomastoideus), а затем передний бугорок поперечного отростка С6 (сонный бугорок).
Рис. 2.2 Центр рабочей поверхности датчика устанавливают над сонным бугорком.
Рис. 2.3 Массивный передний бугорок поперечного отростка С6 (ТА) отбрасывает характерную дорсальную тень. Медиальнее внутренней яремной вены (v. jugularis interna; VJI) и общей сонной артерии (a. carotis communis; ACC) визуализируется щитовидная железа (GT). Под предпозвоночной фасцией (f. cervicalis profunda) проходят длинная мышца шеи (MLCo) и длинная мышца головы (MLCa).
Рис. 2.4 Средний шейный узел (стрелка) визуализируется в области ТОВ (между сосудами и превертебральными мышцами). Маленькой стрелкой обозначен блуждающий нерв (n. vagus).
Содержание книги "Ультразвуковая топографическая анатомия периферической нервной системы"
1 Как извлечь пользу из книги: «Руководство пользователя»!
2 Шея
3 Плечо, предплечье и кисть
4 Туловище
5 Ягодичная область
6 Бедро, голень и стопа
Книга содержит всего пять основных разделов:
- Шея
- Верхняя конечность (плечо, предплечье/кисть)
- Туловище
- Ягодичная область
- Нижняя конечность (бедро, голень/стопа)
Нервы, проходящие в этих анатомических областях, перечисляются в алфавитном порядке (с номерами страниц) на латыни в соответствии с Terminología Anatómica (Международной анатомической терминологией) FCAT (Федеративного международного комитета по анатомической терминологии; 1998 IFFA; FACT; издательство Georg Thieme, Штутгарт, ФРГ) и описываются в таком порядке, каждый нерв на отдельном раз-вороте книги:
- Основные ветви (ramus/rami) необходимо искать по наименованию нерва, дающего им начало!
- Нерву, упоминаемому повторно, присваивается порядковый номер, а также приводится наименование анатомической области!
Пример: ладонная ветвь срединного нерва (r. palmaris n. mediani)
Пошаговый алгоритм поиска соответствующей ветви:
1. Выберите анатомическую область - см. раздел «Предплечье/кисть».
2. Найдите в списке заголовок «Nervus medianus (срединный нерв)».
3. Найдите подзаголовок «Nervus medianus, r. palmaris (ладонная ветвь срединного нерва)» с указанными номерами страниц.
4. Для ознакомления с материалом откройте указанную страницу. и. Руководство пользователя!
Вступление к книге "Ультразвуковая топографическая анатомия периферической нервной системы"
Если мы хотим научиться безошибочно находить и идентифицировать периферические нервы при проведении местной анестезии, лечении болевого синдрома и выполнении других клинических манипуляций, нам необходимы глубокие познания в области клинической анатомии. Ориентироваться при поиске нервов помогают находящиеся рядом с ними крупные сосуды, мышцы, костные образования и другие структуры. Для начинающих специалистов в области эхографии, изучающих ультразвуковую диагностику и анатомию нервной системы, непросто самостоятельно и быстро находить нужные анатомические образования. Этот процесс часто напоминает навигацию по незнакомой местности без карты и компаса. Такое путешествие требует тщательной подготовки, сбора информации и ознакомления с ландшафтом. Это особенно верно для проведения регионарной анестезии под ультразвуковым контролем и прочих интервенционных методов обезболивания или диагностики. Данный иллюстрированный справочник по ультразвуковому исследованию (УЗИ) периферической нервной системы составлен профессорами Hannes Gruber, Alexander Loizides, Bernhard Moriggl и другими экспертами в области УЗИ периферических нервов и служит этаким спасительным путеводителем по неизведанному или своеобразными картами «Google» для ультразвуковой навигации по периферической нервной системе. Справочник написан доступным языком, имеет простую структуру и содержит иллюстрации с основными анатомическими ориентирами, а также практические рекомендации по технике исследования и стандартные алгоритмы поиска нервов. Книга освещает основные и альтернативные алгоритмы поиска нервов с учетом вариабельности анатомии человека, на которую часто ссылается профессор Moriggl, лучший из известных мне специалистов в области ультразвуковой анатомии. Подобный ультразвуковой атлас - отличный гид по малоизученным анатомическим ландшафтам. Я уверен, что эта уникальная книга - бесценный помощник в изучении ультразвуковой диагностики, анатомии опорно-двигательного аппарата, а также периферической нервной системы. К вам на помощь придут глубочайшие познания авторов и их практические советы по технике исследования с пошаговыми алгоритмами поиска отдельных нервов. Справочник, отличающийся лаконичностью и глубиной подачи материала, подготовит вас к путешествию по неизведанным территориям периферической нервной системы. И пусть это издание станет вашей настольной книгой! Vincent W.S. Chan, MD, FRCPC, FRCA Отделение анестезиологии Университет Торонто Торонто, Онтарио, Канада
«Для кого и зачем?» Целевая аудитория и предназначение этой книги. Эта книга задумана как карманный путеводитель для коллег, уже практикующих УЗИ нервов и еще только интересующихся этой методикой. Она позволит быстро найти любые нервы в повседневной клинической практике. Можно выразиться и так: тебе больше не нужно искать нерв, ибо ты его уже нашел в этой книге. Таким образом, ответ на вопрос «зачем?» уже почти готов, ведь подобные руководства еще не издавались. Наша книга позволит сэкономить драгоценное время для диагностики, инвазивных вмешательств и/или планового лечения. Четкие и краткие (!) иллюстрированные описания видимых и/или пальпируемых «внешних» ориентиров позволят быстро установить ультразвуковой датчик в оптимальной исходной позиции. На УЗ-изображениях отмечены лишь основные «внутренние» ориентиры, позволяющие локализовать нерв и охарактеризовать его топографическое местоположение среди других структур. В практических целях мы выделили зоны, где наиболее четко визуализируются отдельные нервы и ветви, назвав их «Точками Оптимальной Видимости» (ТОВ). Практически каждый (!) периферический нерв имеет свою ТОВ, которая приводится в обзорных таблицах по отдельным нервам. Твердое знание ТОВ поможет ориентироваться в сложных ситуациях, особенно при плохом акустическом доступе. Авторы уделили особенное внимание практическим моментам УЗИ периферических нервов, что позволило им описать анатомические варианты прохождения нервов и постараться рассмотреть альтернативные алгоритмы их поиска. Дополняют книгу комментарии по позициям датчика и частым техническим ошибкам. Ссылки на бесчисленные исследования по УЗИ периферических нервов опущены намеренно, так как эти сведения воспрепятствовали бы краткости изложения материала. Мы очень надеемся, что этот справочник придется вам по душе и станет вашей настольной книгой! А завершим мы наше вступительное слово вариацией на тему цитаты известного немецкого писателя Вильгельма Буша (1832-1908): Вы никогда не найдете нерв там, где его никогда не было. Пусть наша книга поможет вам избежать этой ошибки! Инсбрук, Австрия Bernhard Moriggl Alexander Loizides Hannes Gruber
Герпетический стоматит — острое или рецидивирующее воспаление слизистой оболочки полости рта, проявляющееся быстроразвивающимися везикулезными элементами (пузырьками), сопровождающее общую герпетическую инфекцию организма. Особенностью герпетической инфекции является пожизненное носительство вируса после перенесенного заболевания.
Причиной заболевания являются Herpes simplex virus I или II типа, которые проявляют тропизм к ганглиям тройничного нерва и спинномозговым ганглиям области крестцово-подвздошного сочленения соответственно.
Клинически в полости рта герпетическая инфекция проявляется в двух формах.
Острый герпетический стоматит — заболевание вирусной этиологии, возникает как у взрослых, так и у детей. В последнее время острый герпетический стоматит рассматривают как проявление первичной герпетической инфекции вирусом простого герпеса в полости рта. Инкубационный период заболевания продолжается 1-4 сут. Для продромального периода характерно увеличение поднижнечелюстных (в тяжелых случаях — шейных) лимфатических узлов. Температура тела повышается до 37-41 °C, отмечаются слабость, бледность кожных покровов, головная боль и другие симптомы.
В начале заболевания поражения имеют вид пузырьков, которые могут локализоваться в любых участках слизистой оболочки полости рта. Пузырьки быстро вскрываются с образованием поверхностных эрозий, напоминающих афты, которые часто определяются на фоне отечной, гиперемированной, воспаленной слизистой оболочки полости рта. Заболевание контагиозно для лиц, ранее не инфицированных вирусом простого герпеса. Дети в возрасте от 1 года до 3 лет и взрослые в молодом возрасте болеют в основном стоматитом, что составляет 70%. Слизистая оболочка полости рта, особенно десневого края, отечна, гиперемирована. Весь процесс формирования афт длится 4-5 дней. При этом пациент жалуется на боль при приеме пищи, жжение, зуд. Часто заболевание сопровождается увеличением регионарных лимфатических узлов. При недостаточном уходе за полостью рта возможно присоединение бактериальной микрофлоры с развитием более глубоких поражений слизистой оболочки. Если не провести нужного лечения острого герпетического стоматита, возникает рецидивирующая форма, которая сопровождается регулярными высыпаниями на слизистой оболочке полости рта пузырьков и афт.
По степени тяжести различают легкую, среднетяжелую и тяжелую формы.
При легкой форме заболевания симптомы интоксикации организма отсутствуют. Элементы поражения появляются в виде двух-трех небольших пузырьков. После самопроизвольного вскрытия эпителизация слизистой оболочки наступает быстро. Среднетяжелая форма сопровождается выраженной интоксикацией. Появляются множественные высыпания на слизистой оболочке полости рта. При тяжелой форме выражены все признаки острого инфекционного заболевания (высокая температура тела, симптомы интоксикации и др.).
Хронический рецидивирующий герпес является следствием пожизненного носительства вируса и проявляется в виде одиночных высыпаний или тесно расположенных групп мелких пузырьков на красной кайме губ, слизистой оболочке нёба, глаз, половых органов, коже губ, крыльев носа. Обычно появлению этих поражений предшествует чувство жжения, зуда, позже на фоне участков гиперемии возникают пузырьки, которые самопроизвольно вскрываются. Затем элементы сливаются и образуют эрозивные поверхности, и при приеме пищи, разговоре возникают болезненные ощущения.
Диагностика включает использование микроскопического, серологического и молекулярно-биологического (ПЦР) методов. Готовят мазки-отпечатки, окрашенные по Романовскому-Гимзе, в которых обнаруживают характерные многоядерные гигантские клетки. С помощью иммунофлуоресценции можно обнаружить в пораженных клетках вирусный антиген. Серологическим методом (реакцией связывания комплемента) и ИФА выявляют повышение титра антител к вирусу герпеса. Также можно провести выделение вируса, заражая куриный эмбрион или культуру клеток почек кролика (вирусологический метод).
Лечение и профилактика
Применяют противовирусные химиотерапевтические препараты (ацикловир, идоксуридин, изопринозин, теброфеновую и флореналевую мази местно). Для профилактики разработана инактивированная культуральная герпетическая вакцина ВПГ-1/2.
Книга "Микробиология, вирусология, иммунология полости рта. Учебник"
Автор: В. Н. Царев
Издание учебника, переработанное и дополненное в связи с изменением образовательной программы по специальности “Стоматология” согласно стандарту ФГОС 3+, включает сведения об основных этапах становления микробиологии, вопросы специальной терминологии. В нем рассмотрены вопросы таксономии, экологии и биологии микробов, населяющих слизистую оболочку полости рта человека, общие принципы взаимоотношений человека с микроорганизмами.
Изложены физико-химическое обоснование эффективности современных методов стерилизации и дезинфекции применительно к нуждам практической стоматологии, принципы антибактериальной и иммуномодулирующей терапии в стоматологии. Подробно представлен материал по иммунологии полости рта. Обновлено и расширено описание возбудителей основных инфекционных заболеваний человека, вызывающих патологические проявления в полости рта. Отдельные главы учебника посвящены вопросам формирования микробной биопленки, развитию кариеса зубов, заболеваний пародонта, слизистой оболочки полости рта и их осложнениям местного и системного характера.
Учебный материал, иллюстрированный таблицами, схемами, фотографиями и дополненный нормативными документами, изложен авторами в оригинальной и простой концепции, которая помогает студентам усвоить основы общей и специальной микробиологии, иммунологии и вирусологии.
Предисловие к книги "Микробиология, вирусология, иммунология полости рта. Учебник" - В. Н. Царев
Микробиология полости рта — это раздел медицинской микробиологии, включающий закономерности функционирования бактериального сообщества полости рта и организма человека во всем многообразии их взаимодействия между собой и внешним миром микробов. В последние годы изучению так называемой нормальной (резидентной) микробиоты (по-старому — микрофлоры) посвящается все больше и больше исследований. Микробиота полости рта — совокупность разнообразных микроскопически малых живых существ, способных вступать в симбионтные отношения между собой и человеком как биологическим видом.
Микробиота полости рта крайне разнообразна и сложна для изучения. И чем больше мы познаем этот мир, тем больше вопросов возникает у исследователей. Несомненное преобладание представителей бактерий с анаэробным типом дыхания в составе бактериального сообщества полости рта требует специальных условий для культивирования и идентификации этих микроорганизмов, что существенно осложняет диагностику вызываемых ими заболеваний. Не менее сложны и многогранны механизмы неспецифической и иммунной защиты, контролирующие процессы бактериальной колонизации, формирование биопленок и т.п.
Все это определяет необходимость изучения студентами стоматологических факультетов медицинских вузов такого важного раздела, как микробиология, вирусология и иммунология полости рта, что послужило основанием для выделения микробиологии полости рта как отдельной учебной дисциплины. В настоящем учебнике, который представляет собой первое в отечественной практике издание такого рода, изложен материал по микробиологическим аспектам отдельных направлений стоматологии: терапевтической стоматологии, пародонтологии и эндодонтии, хирургической и ортопедической стоматологии, что позволит использовать его для модульного обучения студентов на соответствующих кафедрах стоматологических факультетов медицинских вузов.
Большое внимание в соответствующих разделах учебника уделено особенностям физиологических защитных механизмов и иммунных явлений в полости рта, и особенно — основным принципам антибактериальной и иммуномодулирующей химиотерапии, которым не уделяется достаточного внимания в традиционных учебниках для медицинских вузов.
Авторы настоящего издания стремились обобщить основные сведения о микробиоте полости рта в норме и при патологии, используя как собственный опыт, накопленный на кафедре микробиологии, вирусологии, иммунологии Московского государственного медико-стоматологического университета им. А.И. Евдокимова, так и опыт одноименных кафедр Первого Московского государственного медицинского университета им. И.М. Сеченова (Сеченовского Университета), Омского государственного медицинского университета, Ставропольского государственного медицинского университета, Ростовского государственного медицинского университета, Тверского государственного медицинского университета, Северо-Осетинской государственной медицинской академии, других вузов России, данные современной иностранной литературы.
Все замечания и пожелания в отношении структуры и содержания учебника по микробиологии и вирусологии полости рта, характеру изложения материала и иллюстрациям будут с благодарностью приняты авторами.
В. Н. Царев, доктор медицинских наук, профессор, заслуженный работник высшей школы РФ
Возбудители — дрожжевые грибы рода Candida, в небольшом количестве присутствующие на слизистых оболочках здорового человека. Активизации способствуют врожденные или приобретенные дефекты иммунной системы, гиповитаминозы, эндокринные нарушения, гормональная, лучевая или химиотерапия онкологических заболеваний, иногда — длительная (месяцы) антибактериальная терапия.
Клиническая картина
В последние годы отмечается рост заболеваемости кандидозом. Это связано с широким применением в лечебной практике антибиотиков и особенно иммунодепрессантов — глюкокортикоидов и цитостатиков. Кандидоз — классический пример дисбиоза. Грибы рода Candida являются типичным резидентом слизистых оболочек полости рта, пищеварительной системы, дыхательных путей, влагалища, но в норме определяются в незначительном количестве. Длительное применение антибактериальных препаратов приводит к нарушению состава нормальной микрофлоры. Немаловажную роль в этом процессе играет снижение активности Т-системы и защитных механизмов макрофагально-гранулоцитарной системы организма. Именно поэтому кандидозный стоматит может быть первым проявлением СПИДа.
Рис. 36.1. Кандидоз слизистой оболочки полости рта и языка
Клинические проявления кандидоза полости рта весьма разнообразны. В последние годы, помимо известных форм кандидозного стоматита, встречается Candida-ассоциированный пародонтит, язвенно-эрозивный стоматит, особенно при резком ослаблении защитных сил организма, иммунодефиците, СПИДе. Осложнением процесса является генерализация микоза с развитием Candida-cencnca, нередко с летальным исходом.
Местное поражение полости рта грибами рода Candida (С. abicans, С. tropicalis, С. krusei) называется молочницей, более обширное поражение — кандидозом. Встречаются поражения языка, слизистой оболочки щек, губ, углов рта.
Кандидоз полости рта характеризуется появлением на фоне гиперемированной, сухой и болезненной слизистой оболочки молочно-белого, рыхлого, легко снимаемого налета, после удаления которого обычно обнаруживаются эрозированные участки слизистой оболочки.
Следует также отметить, что Candida часто играют роль участника бактериальных ассоциаций при различных бактериальных инфекциях (хроническом тонзиллите, дифтерии и носительстве дифтерийной палочки, дизентерии). В таких ассоциациях Candida способствуют проявлению патогенности других микроорганизмов, осложняют течение инфекционного заболевания.
Диагностика и лечение
Для лабораторной диагностики используют следующие методы:
микроскопическое исследование (световое, люминесцентное) патологического материала (налета, кусочков органов и т.д.) и обнаружение псевдомицелия (рис. 36.2);
микологическое (культуральное) исследование — посев материала на среды Сабуро, томатный или картофельный агар с последующей идентификацией выделенной культуры;
серологический метод — постановку реакций агглютинации и связывания комплемента с кандидозным антигеном в целях обнаружения антител в сыворотке крови больного в возрастающем титре.
При кандидозах необходимо проводить как этиологическое, так и патогенетическое лечение. В настоящее время более 50% штаммов Candida устойчивы к нистатину и 70-75% — к леворину. Основными противокандидозными средствами являются производные азола — кетоконазол (низорал), флуконазол (дифлюкан, флюкостат), итраконазол (орунгал). При генерализованных процессах применяют амфотерицин В и итраконазол. В случае иммунодефицитов необходимо назначение иммуномодуляторов.
Рис. 36.2. Дрожжевые клетки (бластоспоры) и нити псевдомицелия рода Candida в мазке из материала от больного кандидозом
Книга "Микробиология, вирусология, иммунология полости рта. Учебник"
Автор: В. Н. Царев
Издание учебника, переработанное и дополненное в связи с изменением образовательной программы по специальности “Стоматология” согласно стандарту ФГОС 3+, включает сведения об основных этапах становления микробиологии, вопросы специальной терминологии. В нем рассмотрены вопросы таксономии, экологии и биологии микробов, населяющих слизистую оболочку полости рта человека, общие принципы взаимоотношений человека с микроорганизмами.
Изложены физико-химическое обоснование эффективности современных методов стерилизации и дезинфекции применительно к нуждам практической стоматологии, принципы антибактериальной и иммуномодулирующей терапии в стоматологии. Подробно представлен материал по иммунологии полости рта. Обновлено и расширено описание возбудителей основных инфекционных заболеваний человека, вызывающих патологические проявления в полости рта. Отдельные главы учебника посвящены вопросам формирования микробной биопленки, развитию кариеса зубов, заболеваний пародонта, слизистой оболочки полости рта и их осложнениям местного и системного характера.
Учебный материал, иллюстрированный таблицами, схемами, фотографиями и дополненный нормативными документами, изложен авторами в оригинальной и простой концепции, которая помогает студентам усвоить основы общей и специальной микробиологии, иммунологии и вирусологии.
Предисловие к книги "Микробиология, вирусология, иммунология полости рта. Учебник" - В. Н. Царев
Микробиология полости рта — это раздел медицинской микробиологии, включающий закономерности функционирования бактериального сообщества полости рта и организма человека во всем многообразии их взаимодействия между собой и внешним миром микробов. В последние годы изучению так называемой нормальной (резидентной) микробиоты (по-старому — микрофлоры) посвящается все больше и больше исследований. Микробиота полости рта — совокупность разнообразных микроскопически малых живых существ, способных вступать в симбионтные отношения между собой и человеком как биологическим видом.
Микробиота полости рта крайне разнообразна и сложна для изучения. И чем больше мы познаем этот мир, тем больше вопросов возникает у исследователей. Несомненное преобладание представителей бактерий с анаэробным типом дыхания в составе бактериального сообщества полости рта требует специальных условий для культивирования и идентификации этих микроорганизмов, что существенно осложняет диагностику вызываемых ими заболеваний. Не менее сложны и многогранны механизмы неспецифической и иммунной защиты, контролирующие процессы бактериальной колонизации, формирование биопленок и т.п.
Все это определяет необходимость изучения студентами стоматологических факультетов медицинских вузов такого важного раздела, как микробиология, вирусология и иммунология полости рта, что послужило основанием для выделения микробиологии полости рта как отдельной учебной дисциплины. В настоящем учебнике, который представляет собой первое в отечественной практике издание такого рода, изложен материал по микробиологическим аспектам отдельных направлений стоматологии: терапевтической стоматологии, пародонтологии и эндодонтии, хирургической и ортопедической стоматологии, что позволит использовать его для модульного обучения студентов на соответствующих кафедрах стоматологических факультетов медицинских вузов.
Большое внимание в соответствующих разделах учебника уделено особенностям физиологических защитных механизмов и иммунных явлений в полости рта, и особенно — основным принципам антибактериальной и иммуномодулирующей химиотерапии, которым не уделяется достаточного внимания в традиционных учебниках для медицинских вузов.
Авторы настоящего издания стремились обобщить основные сведения о микробиоте полости рта в норме и при патологии, используя как собственный опыт, накопленный на кафедре микробиологии, вирусологии, иммунологии Московского государственного медико-стоматологического университета им. А.И. Евдокимова, так и опыт одноименных кафедр Первого Московского государственного медицинского университета им. И.М. Сеченова (Сеченовского Университета), Омского государственного медицинского университета, Ставропольского государственного медицинского университета, Ростовского государственного медицинского университета, Тверского государственного медицинского университета, Северо-Осетинской государственной медицинской академии, других вузов России, данные современной иностранной литературы.
Все замечания и пожелания в отношении структуры и содержания учебника по микробиологии и вирусологии полости рта, характеру изложения материала и иллюстрациям будут с благодарностью приняты авторами.
В. Н. Царев, доктор медицинских наук, профессор, заслуженный работник высшей школы РФ
Проблема обильных менструальных кровотечений в практике врача-гинеколога остается одной из нерешенных. Точная частота встречаемости обильных менструальных кровотечений неизвестна. Анализу информации мешают различия в культурном, социальном и возрастном восприятии «нормальной» менструации. Обильные менструальные кровотечения считаются наиболее распространенным типом аномальных маточных кровотечений (АМК). Чрезмерная кровопотеря во время менструации оказывает отрицательное влияние на физическое, социальное, эмоциональное (анемия, болевой синдром, чувство усталости, изменение качества жизни) и/или материальное благополучие женщины и ее семьи. У женщин с тяжелыми формами обильных менструальных кровотечений риски госпитализации повышаются в 2,7 раза, вызовов скорой помощи - в 1,4 раза, амбулаторных посещений врача - в 1,3 раза по сравнению с женщинами без подобных жалоб.
Согласно статистическим данным (Narice B.F. et al., 2018), в Великобритании ежегодно за медицинской помощью в связи с аномальными маточными кровотечениями (АМК) обращаются 1 млн женщин; по поводу аномальных маточных кровотечений было выполнено в среднем 17,8 оперативного вмешательства на 10 000 женщин репродуктивного возраста в США. При этом в 2007 г. общие годовые прямые и косвенные затраты на АМК в США превысили 37 млрд долларов. В РФ аномальные маточные кровотечения встречается хотя бы 1 раз в жизни более чем у половины женщин репродуктивного возраста. Пристальное внимание обращено к АМК в настоящий момент и потому, что, наряду с возрастом старше 60 лет, СД, системной АГ, ожирением, приемом тамоксифена, аномальные маточные кровотечения связаны со злокачественными новообразованиями эндометрия.
Поданным FIGO (Munro M.G. et al., 2018), почти 50% женщин с аномальными маточными кровотечениями не обращаются за медицинской помощью. Это, во-первых, влияет на частоту выявления патологии, во-вторых, увеличивает частоту материнской заболеваемости и смертности среди беременных женщин с анемией, связанной с аномальными маточными кровотечениями.
В сознании женщины наличие менструации обозначает фертильность и отсутствие беременности. При проведении эпидемиологических исследований (п=475) был показан низкий уровень осведомленности женской популяции и выявлены проблемы понимания обильных менструальных кровотечений. Было показано, что 41% женщин с кровопотерей более 80 мл оценивали свои менструации как необильные или даже скудные, а 14% женщин, теряющих менее 20 мл менструальной крови, считали свои менструации обильными. Недостаточная информированность и непонимание проблемы ограничивает обращение женщин за помощью. В настоящее время нет точных данных о достоверном количестве женщин с обильными менструальными кровотечениями.
Чтобы адаптироваться к обильным менструальным кровотечениям, женщины (более 50%) «подстраивают» свою жизнедеятельность под менструации: избегают сексуальной активности, носят специальное белье или одежду, используют несколько средств защиты одновременно, избегают длительного сидения и активных мероприятий в эти дни. В исследовании было показано, что социальная активность (учеба или работа, занятия спортом и домашние дела) и сексуальная жизнь нарушены более чем у 2/3 женщин (п=1627) с обильными менструальными кровотечениями. Сама женщина с аномальными маточными кровотечениями или обильными менструальными кровотечениями сообщает о значительном снижении качества жизни. Но, с другой стороны, согласно данным исследования NICE (National Institute for Health and Care Excellence, Великобритания), только 18% врачей (при этом нет зависимости от возраста, пола, опыта, места работы врача) при анонимном опросе считают изменение качества жизни важным аспектом при обсуждении проблемы аномальных маточных кровотечений с пациенткой (Lam С. et al., 2017), что абсолютно противоречит даже определению аномальных маточных кровотечений.
Определение обильных менструальных кровотечений, предложенное NICE в 2007 г., в наибольшей степени подходит для клинической практики и является альтернативой определению, основанному на физическом измерении кровопотери, что, как показывает практика, является очень субъективным показателем. Согласно NICE, обильные менструальные кровотечения - это чрезмерная менструальная кровопотеря, которая оказывает влияние на физическое, социальное, эмоциональное и/или материальное благополучие женщины. Данное определение полностью было подтверждено в документе NICE в 2018 г и введено в терминологию нормального и аномального маточного кровотечения FIG0 (International Federation of Gynecology and Obstetrics) в этом же году. В нашей клинической практике мы окончательно перестали использовать в лексике термины «меноррагия», «гиперменорея», «гипоменорея», «менометроррагия», «дисфункциональное маточное кровотечение» - существуют только аномальные маточные кровотечения, обильные менструальные кровотечения, обильное и длительное менструальное кровотечение, межменструальное кровотечение.
Рис. 2.6. Классификационная система FIGO причин аномальных маточных кровотечений у небеременных женщин репродуктивного возраста (PALM-COEIN) (2011).
Причинами обильных менструальных кровотечений могут быть как органическая патология, так и функциональная. В 2011 г. впервые была принята «Классификационная система FIGO причин АМК у небеременных женщин репродуктивного возраста (PALM-COEIN)» (рис. 2.6). В ней представлены 9 основных категорий: polyps (полипы) (АМК-Р); adenomyosis (аденомиоз) (АМК-А); leiomyoma (лейомиома) (AMK-L); malignancy (малигнизация) (АМК-М) и hyperplasia (гиперплазия); coagulopathy (коагулопатия) (АМК-С); ovulatory dysfunction (овуляторная дисфункция) (АМК-О); endometrial (эндометриальное) (АМК-Е); iatrogenic (ятрогенное) (AMK-I) и not yet classified (еще не классифицированное) (AMK-N).
В 2018 г. эксперты FIGO изменили некоторые позиции в терминологии для нормального и аномального маточных кровотечений.
Частота:
аменорея.
Регулярность:
нормальное изменение - 8 дней (разница между самым коротким и самым длинным менструальными циклами);
незначительная разница в зависимости от возраста.
Длительность:
нормальная — до 8 дней;
увеличенная - более 8 дней.
Объем и определение симптомов обильных менструальных кровотечений:
определение NICE;
объем кровотечений, достаточный для того, чтобы негативно повлиять на качество жизни женщины.
Межменструальное кровотечение:
спонтанное кровотечение, происходящее между менструациями;
циклическое;
беспорядочное.
Также была проведена коррекция в отношении классификационной системы PALM-COEIN:
2. AMK-L. Определение 3-го типа узла как субмукозной миомы матки. Типы и различия между узлами:
Типы 0 и 1; 6 и 7.
Типы 2 и 3; 4 и 5.
3. АМК-С. Больше не включает АМК, связанные с применением препаратов, изменяющих свертывание крови: теперь они включены в AMK-I.
4. АМК-I. Включает АМК, связанные с любой ятрогенией, в том числе с применением антикоагулянтов и препаратов, влияющих на овуляцию.
5. АМК-О. Введены новые диагностические пороговые изменения характеристик нормального кровотечения и цикла, описанные ранее. Не включает овуляторные расстройства, связанные с применением лекарственных средств, достоверно или предположительно влияющих на овуляцию.
6. AMK-N. Название категории было изменено с «еще не классифицированы» на «не относятся ни к одной из категорий».
Книга "Эндокринная гинекология: избранные семинары"
Авторы: Е. Н. Андреева, О. Р. Григорян
В настоящем издании собраны основные аспекты эндокринной гинекологии, с которыми гинеколог и/или эндокринолог встречаются на своем амбулаторном приеме.
Комплексный подход к решению этих проблем крайне важен, поскольку без знаний эндокринной гинекологии невозможно оказать пациентке полноценную помощь. Акушер-гинеколог, ведущий амбулаторный прием в женской консультации, сталкивается с необходимостью наблюдать женщину в различные периоды ее жизни. Знание основ эндокринной гинекологии позволит врачу первичного звена выявлять патологии на ранних стадиях, а главное, оказывать адекватную медицинскую помощь с учетом сопутствующих заболеваний, имеющихся у пациентки.
Рассмотрены современные данные о патогенезе и клинической картине эндокринных нарушений в зависимости от возрастных аспектов, представлены методы диагностики, актуальные принципы и методы терапии различной патологии эндокринной и репродуктивной систем. Даны основы подбора контрацепции и менопаузальной гормональной терапии с учетом сопутствующих эндокринных расстройств.
Нарушения менструального цикла. Этиологические и патогенетические факторы дисфункции яичников у женщин репродуктивного возраста
Нарушения менструального цикла (МЦ) - одно из частых проявлений (а иногда - причина) гинекологических заболеваний у женщин. Несмотря на большие адаптационные возможности женского организма, в последние 30 лет отмечается неуклонный рост нарушений репродуктивной функции. По данным Е. А. Куксиной (2016), число женщин с расстройством менструации с 1980-го по 2016 г. увеличилось в 7,3 раза, а средний возраст женщин с нарушениями менструального цикла составил 29,8 года. Следует подчеркнуть, что данный возраст является «расцветом» репродуктивного периода, когда наши пациентки планируют рождение первого ребенка. Средний возраст матери при рождении первенца продолжает медленно расти, и в 2017 г. он составил 28,5 года. На сегодняшний день Правительство РФ ставит демографическую задачу увеличения численности населения, поэтому врач-клиницист обращает особенное внимание на репродуктивный период женщины, поскольку нарушения в этот период могут препятствовать наступлению беременности и приводить к бесплодию. Было замечено, что у девушек, проживающих в сельской местности, в 1,5 раза реже наблюдаются нарушения менструального цикла по сравнению с городскими сверстницами. Можно предположить, что это связано с выраженными стрессовыми факторами, с которыми городские жительницы сталкиваются чаще, чем проживающие в сельской местности, а также с нарушением циркадных ритмов и снижением синтеза мелатонина у жительниц больших городов. Интересный факт был отмечен Т. А. Густоваровой и соавт. (2014): девушки, рожденные и воспитываемые во внебрачных семьях, имели нарушения менструального цикла с частотой 28,1% по сравнению с 6,3% среди девочек из полных семей.
Одной из основных проблем ведения женщин в репродуктивном периоде является недостаточная просветительская помощь данной категории населения. Некоторые наши пациентки и их родственницы (мамы, бабушки, тети) не считают это проблемой и, следовательно, не обращаются за медицинским консультированием при нарушениях менструального цикла и ненаступлении беременности. Задача гинеколога во время консультирования - донесение информации о том, что регулярный менструального цикла является маркером физического и психического благополучия женщины репродуктивного возраста. Однако одной из главных сложностей для медицинского персонала при ведении женщин, в том числе репродуктивного возраста, является дефицит времени, - этот фактор отмечают 86% врачей. Также 68% врачей указали на проблему недоверия со стороны женщин. Согласно работе A.Lania и соавт. (2019), врач на приеме, как правило, сразу, не собирая анамнез, назначает обследования женщине с нарушениями менструального цикла и далее «лечит» ее нарушения, выявленные при обследовании (например, гормональные), не воздействуя на патогенетический фактор этих нарушений. Задачей клинициста, согласно рекомендациям ВОЗ, является охрана репродуктивного здоровья, которое включает репродуктивные процессы, функции и систему на всех этапах жизни, даже в те периоды, когда пациентка не планирует беременность.
Многообразие нозологических форм нарушений менструального цикла обусловлено его многоступенчатой регуляцией. Нарушения могут быть следствием как непосредственной дисфункции гипоталамо-гипофизарно-яичниковой оси (рис. 2.1), так и влияния на нее извне (например, стресса, нарушений пищевого поведения, неадекватной физической нагрузки и т.д.). Нейро гуморальная регуляция менструальной функции осуществляется вследствие согласованной работы коры больших полушарий, специфических отделов гипоталамуса, гипофиза, а также их взаимодействия с периферическими эндокринными органами, рядом экстрагипоталамических структур. Как правило, нарушения менструального цикла связаны с изменениями в системе регуляции репродуктивной функции или в органах-мишенях. Важным открытием стали идентификация и установление функции кисспептина в половой дифференциации головного мозга, секреции гонадотропинов и индукции овуляции. Нейроны, экспрессирующие кисспептин (KISSI- нейроны), можно считать медиаторами между энергетическим балансом и репродуктивной системой. Кисспептин усиливает секрецию Л Г и ФСГ как у женщин, так и у мужчин. Стимулирующее действие кисспептина на гипоталамо-гипофизарно-яичниковую ось было выявлено при различных физиологических состояниях, в разные фазы менструального цикла, а также при беременности и лактации.
Рис. 2.1. Звенья (центральные и периферические) регуляции женской репродуктивной системы.
ГнИГ- гонадотропинингибирующий гормон.
Кисспептин синтезируется во многих органах: в гипоталамусе, плаценте, гонадах, почках, поджелудочной железе, печени, кишечнике, аорте, венечных артериях и пупочной вене. Экспрессия кисспептина и его рецептора в гипоталамусе происходит в основном в нейронах аркуатного ядра и антеровентрального перивентрикулярного ядра преоптической области.
Общепринятая терминология, к сожалению, не позволяет достичь взаимопонимания между специалистами в вопросах диагностики и лечения заболевания, которое в Международной классификации болезней 10-го пересмотра носит название «дисфункция яичников» (Е28). Данный термин, по сути, является первичным диагнозом, который после осмотра, сбора анамнеза, дообследования будет отражать характер и причину нарушений менструального цикла, а следовательно, каждая пациентка получит индивидуальный подход к своей проблеме.
Основные «негормональные» и «отличные от органических» причины нарушений регуляции репродуктивной системы включают стрессовый фактор, колебания массы тела, нарушения пищевого поведения, физическую нагрузку без учета индивидуальных особенностей, прием лекарственных препаратов, оказывающих влияние на синтез, метаболизм, рецепцию и обратный захват нейротрансмиттеров и нейромодуляторов. Важно помнить, что одно и то же нарушение менструального цикла может быть вызвано различными факторами, и одна и та же причина может приводить к формированию различных нарушений менструального цикла. При длительном существовании провоцирующего фактора (например, стресса или колебания массы тела) формируются нарушения менструального цикла, в которые поэтапно вовлекаются все звенья регуляции, и возникают стабильные нарушения работы гипоталамо-гипофизарно-яичниковой оси.
В 2018 г. экспертная группа FIGO (International Federation of Gynecology and Obstetrics - Международной федерации гинекологии и акушерства) (Munro M.G. et al., 2018), в состав которой вошли представители 17 стран мира, предложила характеристики нормального, регулярного менструального цикла:
• частота (интервал между кровотечениями): 24-38 дней;
• регулярность (интервалы без кровотечений >20 дней в течение 90-дневного периода);
• продолжительность кровотечения: 4,5 - 9 дней.
В качестве параметров нормального менструального цикла выступают не только его продолжительность 24-38 дней, но и наличие овуляции и полноценная лютеиновая фаза. При этом продолжительность цикла зависит от длительности фолликулярной фазы, тогда как длительность нормальной лютеиновой фазы - величина сравнительно постоянная, которая должна составлять около 14 дней.
Следует подчеркнуть, что, согласно рекомендациям последних лет, скрининговой оценке у женщины репродуктивного возраста с нарушениями менструального цикла подлежат плазменные уровни таких гормонов, как ЛГ, ФСГ, эстрадиол, антимюллеров гормон, тестостерон, ДГЭА-С, 17-гидроксипрогестерон, ТТГ, св.Т4, пролактин. Кровь на анализ рекомендуется сдавать натощак в утреннее время в раннюю фолликулярную фазу менструального цикла (самостоятельного или индуцированного) или на фоне отсутствия менструального цикла.
Книга "Эндокринная гинекология: избранные семинары"
Авторы: Е. Н. Андреева, О. Р. Григорян
В настоящем издании собраны основные аспекты эндокринной гинекологии, с которыми гинеколог и/или эндокринолог встречаются на своем амбулаторном приеме.
Комплексный подход к решению этих проблем крайне важен, поскольку без знаний эндокринной гинекологии невозможно оказать пациентке полноценную помощь. Акушер-гинеколог, ведущий амбулаторный прием в женской консультации, сталкивается с необходимостью наблюдать женщину в различные периоды ее жизни. Знание основ эндокринной гинекологии позволит врачу первичного звена выявлять патологии на ранних стадиях, а главное, оказывать адекватную медицинскую помощь с учетом сопутствующих заболеваний, имеющихся у пациентки.
Рассмотрены современные данные о патогенезе и клинической картине эндокринных нарушений в зависимости от возрастных аспектов, представлены методы диагностики, актуальные принципы и методы терапии различной патологии эндокринной и репродуктивной систем. Даны основы подбора контрацепции и менопаузальной гормональной терапии с учетом сопутствующих эндокринных расстройств.
Аменорея - отсутствие или прекращение менструаций. Она является не самостоятельным заболеванием, а симптомом патологии на конкретных уровнях репродуктивной системы, нейроэндокринных заболеваний, доброкачественных и злокачественных новообразований различных органов.
Аменорея подразделяется на первичную и вторичную. Первичная аменорея - отсутствие менструаций в 15 лет (при условии развития вторичных половых признаков) или через 3 года после телархе, а также отсутствие развития вторичных половых признаков и менструаций к возрасту 13 лет. Вторичная аменорея - отсутствие менструаций в течение 6 мес. при ранее нерегулярном менструальном цикле, отсутствие менструаций в течение 3 мес. при ранее регулярном менструальном цикле (МЦ).
Олигоменорея — это нарушение менструального цикла (МЦ), при котором его длительность составляет более 35 дней; или частота менструаций менее 9 в год. Длительность нормального цикла не должна превышать 38 дней, так как данному требованию соответствуют параметры цикла 95% здоровых женщин.
Распространенность аменореи в популяции женщин репродуктивного возраста составляет примерно 1,8-3,5%, среди студенток - 3,5-5%, а в структуре нарушений менструальной и генеративной функции - 10- 15%. Первичная аменорея встречается гораздо реже, чем вторичная, и составляет около 10% в структуре всех форм аменореи.
Причины первичной и вторичной аменореи следующие:
Первичная аменорея:
Первичная аменорея с задержкой полового развития:
пороки развития гонад (дисгенезия гонад);
нарушения функции гипоталамо-гипофизарной системы;
конституциональная форма задержки полового развития;
функциональные нарушения гипоталамо-гипофизарной системы;
органические нарушения гипоталамо-гипофизарной системы.
Первичная аменорея без задержки полового развития:
пороки развития половых органов;
гинатрезия;
аплазия матки.
Вторичная аменорея:
Патология матки:
атрезия цервикального канала;
синдром Ашермана (внутриматочные синехии).
Функциональные нарушения гипоталамо-гипофизарной системы:
аменорея на фоне потери массы тела;
психогенная аменорея;
гиперпролактинемия.
Яичниковые формы аменореи:
преждевременная недостаточность яичников;
синдром гиперторможения гонадотропной функции.
Эндокринные причины:
СПЯ;
ВДКН;
гиперкортицизм;
нарушения тиреоидной функции;
акромегалия и др.
Первичная аменорея
Первичная аменорея - отсутствие менструаций в 15 лет (при условии развития вторичных половых признаков) или через 3 года после телархе, а также отсутствие развития вторичных половых признаков и менструаций к возрасту 13 лет. Выделяют первичную аменорею с нарушением развития вторичных половых признаков и без такового. С точки зрения этиотропной классификации, различают первичную аменорею центрального и яичникового генеза. Для первой характерно резкое снижение уровня гонадотропинов с последующей блокадой стероидогенеза в яичниках (гипогонадотропный гипогонадизм). Первичную аменорею яичникового генеза, сопровождающуюся избытком гонадотропинов, называют гипергонадотропной. Помимо указанных заболеваний, первичная аменорея встречается при ВДКН. Критерии гипогонадизма в зависимости от уровня гонадотропинов: гипергонадотропный (ФСГ >30 МЕ/л), нормогонадотропный (ФСГ 1,6-11 МЕ/л) и гипогонадотропный (ФСГ <1,6 МЕ/л).
Первичная гипогонадотропная аменорея встречается при синдроме Каллманна. Еще одним характерным признаком данной генетической патологии является аносмия. В основе патогенеза синдрома Каллманна, наследуемого аутосомно-рецессивно, лежит мутация гена KALI в Х-хромосоме.
Если первичная аменорея манифестирует на фоне нарушения формирования пола, то перед инициацией пубертата обязательным исследованием является кариотипирование. При нормальном женском кариотипе 46ХХ проводится инициация пубертата с помощью монотерапии эстрогенами в начальной дозе 0,25-0,5 мг эстрадиола с постепенным увеличением дозы на 0,5 мг каждые 6-12 месяцев под контролем УЗИ малого таза. При появлении менструальноподобной реакции дополнительно назначают гестагены или комбинированные эстроген-гестагенные препараты длительно, как минимум до возраста естественной менопаузы. Терапия проводится под контролем врача с обязательным ежегодным обследованием (осмотр гинеколога, УЗИ малого таза, по показаниям УЗИ молочных желез/маммография, мазок на онкоцитологию). При обнаружении Y-хромосомы на первом этапе выполняется гонадэктомия, после чего вторым этапом назначают терапию эстрогенами, при исключении гонадобластомы по данным гистологического заключения.
Пороки развития половых органов
Гинатрезия, атрезия части влагалища - это отсутствие девственной плевы и нижней трети влагалища. Данный порок развития возникает в результате нарушения канализации нижнего отдела урогенитального синуса, из которого формируется нижняя треть влагалища в период внутриутробного развития. Причины изучены недостаточно. Важно помнить, что у 40% пациенток имеются пороки мочевыделительной системы. При атрезии гимена или части влагалища пациентки жалуются на циклические боли внизу живота, что связано с нарушением оттока менструальной крови и формированием гематокольпоса и гематометры (скопление крови в верхней части влагалища и матке). Других жалоб пациентки не предъявляют. Диагностика трудности не представляет. Частота перенесенных заболеваний в анамнезе у таких пациенток не выше, чем в популяции. При физикальном исследовании отмечаются женский морфотип, нормальное развитие вторичных половых признаков. При гинекологическом исследовании находят атрезию гимена или слепо заканчивающееся влагалище.
Концентрации гормонов в крови соответствуют возрастной норме. При УЗИ определяются яичники нормальных размеров и увеличенная матка с расширенной полостью (гематометра). Лечение хирургическое и сводится к рассечению гимена или перегородки во влагалище, что нормализует менструальную и, как следствие, репродуктивную функцию.
Аплазия матки (синдром Майера-Рокитанского—Кюстера) - это отсутствие матки, часто сочетающееся с отсутствием влагалища. Точно установлено, что в яичниках происходят нормальные фолликулогенез, синтез стероидов, овуляция и образование желтого тела. Поэтому нарушения полового развития при этом синдроме отсутствуют: пациентку беспокоит только аменорея.
При физикальном исследовании устанавливают нормальное развитие вторичных половых признаков, женский морфотип. Диагностика аплазии влагалища и матки при гинекологическом исследовании трудности не представляет. При аплазии только матки имеется нижняя треть слепо заканчивающегося влагалища, что подтверждают при вагиноскопии (у девственниц). Гормональные исследования малоинформативны: уровни гонадотропинов и половых стероидов находятся в пределах возрастных нормативов и циклических колебаний. Проба с гестагенами, гестагенами и эстрогенами дает отрицательный результат, что подтверждает маточную форму аменореи. УЗИ позволяет окончательно подтвердить диагноз, при этом определяются яичники нормальных размеров и отсутствие матки.
Лечение хирургическое - кольпопоэз из тазовой брюшины вагинальным доступом параллельно с лапароскопией. После пластической операции возможна половая жизнь. Гормональная терапия не показана ввиду нормальной функции яичников. Необходим ультразвуковой контроль за состоянием яичников, поскольку при отсутствии матки в них часто формируются функциональные кисты. Своевременная антигонадотропная терапия (комбинированные оральные контрацептивы в пролонгированном режиме) способствует их регрессу. Генеративная функция может быть выполнена с помощью вспомогательных репродуктивных технологий в рамках программы суррогатного материнства.
Вторичная аменорея
Вторичная аменорея - это отсутствие менструации в течение 6 мес. и более у ранее менструирующих женщин. При этой форме аменореи нарушение развития вторичных половых признаков отсутствует, поскольку пубертатный период протекает в соответствующих возрастных пределах.
Одной из частых причин вторичной аменореи является дисфункция гипоталамуса на фоне резкой потери массы тела или ограничительных диет, тяжелых физических или психоэмоциональных нагрузок. Эти события приводят к развитию так называемой функциональной гипоталамической аменореи, факт обратимости которой отражен в ее названии. При устранении пускового фактора менструальная функция обычно восстанавливается.
Более редкими причинами вторичной аменореи являются опухоли гипоталамо-гипофизарной области, синдром «пустого» турецкого седла, синдром Шихена, некроз или кровоизлияние в область гипофиза, аменорея на фоне гиперпролактинемии.
В отличие от первичной, вторичная аменорея встречается часто и составляет до 75% случаев в структуре всех форм аменореи. Это частый симптом СПЯ, метаболического синдрома, нарушений функции надпочечников, щитовидной железы. На долю вторичной аменореи при названных нейроэндокринных синдромах приходится более 50% случаев.
Ниже разбираются остальные варианты вторичной аменореи, возникающие в результате травм, заболеваний матки и функциональных нарушений гипоталамо-гипофизарной системы.
Атрезия цервикального канала - отсутствие менструаций в результате травматических внутриматочных манипуляций, выскабливаний, при которых повреждается базальная мембрана эндоцервикса. Частота данной формы аменореи в структуре вторичной аменореи составляет примерно 5-7%. Данная форма аменореи возникает после абортов, выскабливаний, электроконизации шейки матки. В результате травмы разрушается слизистая оболочка цервикального канала до базальной мембраны, активируются факторы адгезии, что приводит к спаечному процессу. Клиническая картина характеризуется отсутствием менструации после хирургических вмешательств, перечисленных выше. Важный симптом — циклические боли вследствие нарушения оттока менструальной крови.
Диагноз устанавливают на основании анамнеза (предшествующий аборт и т.д.), клинической картины и данных УЗИ, произведенного при болях: выявляют расширение полости матки и скопление в ней жидкости (гематометра).
Гормональные исследования неинформативны, поскольку функция яичников не нарушена. Результаты проб с гестагенами, эстрогенами и гестагенами отрицательные, что указывает на маточный уровень поражения репродуктивной системы. Восстановление проходимости цервикального канала путем зондирования можно проводить амбулаторно только при своевременной диагностике. При аменорее давностью более 6-12 мес. показана гистерорезектоскопия.
Внутриматочные синехии (синдром Ашермана) — следствие частых, грубых выскабливаний или эндометритов. Частота заболевания в структуре причин вторичной аменореи составляет примерно 3%. Подобно атрезии цервикального канала, синдром Ашермана нередко развивается после резектоскопии больших субмукозных миом на широком основании, а также после абортов. В отличие от атрезии цервикального канала, циклических болей пациентки не отмечают. Частая ошибка практикующих врачей - поиск эндокринных нарушений при аменорее после аборта. Нет ни одного нейроэндокринного синдрома, который был бы причиной аменореи после аборта или диагностического выскабливания.
Диагностика основана на данных анамнеза - отсутствии менструации после различных хирургических вмешательств. Уровни половых и гонадотропных гормонов находятся в пределах нормы, поэтому эту форму аменореи называют нормогонадотропной. Внутриматочные синехии можно заподозрить по данным трансвагинального УЗИ. Важное диагностическое значение имеет отрицательный результат проб с гестагенами или эстрогенами и гестагенами. При гистероскопии и гистеросальпингографии выявляют типичную картину внутриматочных синехий.
Книга "Эндокринная гинекология: избранные семинары"
Авторы: Е. Н. Андреева, О. Р. Григорян
В настоящем издании собраны основные аспекты эндокринной гинекологии, с которыми гинеколог и/или эндокринолог встречаются на своем амбулаторном приеме.
Комплексный подход к решению этих проблем крайне важен, поскольку без знаний эндокринной гинекологии невозможно оказать пациентке полноценную помощь. Акушер-гинеколог, ведущий амбулаторный прием в женской консультации, сталкивается с необходимостью наблюдать женщину в различные периоды ее жизни. Знание основ эндокринной гинекологии позволит врачу первичного звена выявлять патологии на ранних стадиях, а главное, оказывать адекватную медицинскую помощь с учетом сопутствующих заболеваний, имеющихся у пациентки.
Рассмотрены современные данные о патогенезе и клинической картине эндокринных нарушений в зависимости от возрастных аспектов, представлены методы диагностики, актуальные принципы и методы терапии различной патологии эндокринной и репродуктивной систем. Даны основы подбора контрацепции и менопаузальной гормональной терапии с учетом сопутствующих эндокринных расстройств.
(УЗИ шаблон (пример, бланк) протокола ультразвукового описания патологии печени)
Печень - границы печени не расширены: нижний край правой доли у рёберной дуги, незначительно закруглён, переднезадний размер правой доли 119 мм, косой вертикальный 148 мм, переднезадний размер левой доли 59 мм, вертикальный 93 мм; контуры ровные, диафрагмальный контур нечёткий, паренхима неоднородная за счет гипоэхогенного участка вблизи правой боковой стенки желчного пузыря, имеющего округлую форму, диаметром 15 мм, имеющего ровные, относительно четкие контуры, однородное внутреннее строение; на остальном протяжении паренхима однородная, эхоструктура диффузно повышенной эхогенности рисунок зернистости нечёткий; внутрипечёночные жёлчные протоки не расширены, свободны, сосудистый рисунок обеднен.
Общий жёлчный проток диаметром 4 мм, стенки не утолщены, просвет свободный.
Регионарные лимфатические узлы не визуализируются.
Книга "Практическое руководство по ультразвуковой диагностике. Общая ультразвуковая диагностика" - В. В. Митьков
Фундаментальное клиническое руководство подготовлено коллективом ведущих специалистов ультразвуковой диагностики. В книге представлены разделы, посвященные ультразвуковым диагностическим системам, физическим принципам ультразвуковой диагностики, ультразвуковой диагностике заболеваний печени, желчевыводящей системы, поджелудочной железы, пищевода, желудка, кишечника, селезенки, почек, мочевого пузыря, предстательной железы и семенных пузырьков, надпочечников, органов мошонки, лимфатической системы, молочных, щитовидной, околощитовидных и слюнных желез, органов грудной клетки. Книга предназначена для врачей ультразвуковой диагностики, рентгенологов, радиологов, терапевтов, гастроэнтерологов, эндокринологов, хирургов, урологов, и всех заинтересованных специалистов.
В 3 издании монографии «Эхография в гинекологии» рассмотрены все основные вопросы ультразвуковой диагностики в гинекологии, с которыми ежедневно сталкивается врач, обследующий органы малого таза у женщин в амбулаторной практике и гинекологическом стационаре. Внесены дополнения результатов собственных научных исследований, а также опыта работы ведущих лабораторий мира и нашей страны за последнее время. Особое внимание уделено вопросам стандартизации при обследовании миометрия, эндометрия и яичников, основанных на рекомендациях групп международных экспертов. Написаны новые главы, посвященные послеродовому периоду в норме и при осложнениях, ультразвуковому мониторингу при проведении аборта как медикаментозного, так и путем вакуум-аспирации, а также послеабортным и послеоперационным осложнениям, включая проблему рубца на матке. Каждая глава состоит из небольшого этио-патогенетического раздела, подробно освещены вопросы эхографической диагностики, включая данные цветового картирования, допплерометрии, новых, недостаточно распространённых методик и дифференциально-диагностические критерии. Каждая глава иллюстрирована большим количеством эхограмм как типичного, так и нетипичного изображения рассматриваемой патологии. Определены диагностические возможности эхографии, цветового картирования и допплерометрии во всех рассматриваемых разделах гинекологии. Представлены новые направления диагностики и лечения, внедряемые в гинекологическую практику в течение последних лет. В приложение включены таблицы всех нормативных параметров, предложены протоколы ультразвукового исследования органов малого таза и проведения эхогистеросальпингоскопии. Книга рассчитана на врачей ультразвуковой диагностики, гинекологов, акушеров, онкогинекологов, хирургов и врачей смежных специальностей.
Книга "Эхокардиография от Рыбаковой" - М. К. Рыбакова, В. В. Митьков
Данное издание представляет собой практическое руководство по эхокардиографии, в котором отражены все современные технологии, применяемые в настоящее время. Исключительный интерес для специалистов представляет CD-ROM с подборкой видеоклипов по всем основным разделам, включающих редкие случаи диагностики.
Особенность издания - попытка объединить и сравнить результаты эхокардиографического исследования сердца и паталогоанатомический материал по всем основным разделам. Большой интерес представляют разделы, содержащие новые технологии исследования, такие как трех- и четырехмерная реконструкция сердца в реальном времени, тканевая допплерография. Большое внимание уделено также классическим разделам эхокардиографии – оценке легочной гипертензии, клапанных пороков сердца, ишемической болезни сердца и ее осложнений и т.д.
В книге представлены огромный иллюстративный материал, большое количество схем и рисунков, приведены алгоритмы тактики проведения исследования и диагностики по всем разделам эхоКГ. Руководство помогает разрешить спорные и злободневные вопросы, позволяет ориентироваться в расчетах и измерениях, содержит необходимую справочную информацию. Книга написана сотрудниками кафедры ультразвуковой диагностики ГБОУ ДПО «Российская медицинская академия последипломного образования'' Министерства здравоохранения Российской Федерации» (база – ГКБ им. С.П. Боткина, Москва).
Издание предназначено для специалистов эхокардиографии, врачей ультразвуковой и функциональной диагностики, кардиологов и терапевтов.
Книга "УЗИ в акушерстве и гинекологии. Том 1 Акушерство" - Мерц Эберхард
Данное издание является наиболее полным иллюстрированным руководством по ультразвуковой диагностике в акушерстве и гинекологии.
Авторы постарались охватить все известные на сегодняшний день режимы УЗИ, но тем не менее основной акцент делается на наиболее доступных методиках, технике, глубоком анализе результатов. Результаты ультразвуковых исследований приводятся в тесной корреляции с клиническими и лабораторными данными, что значительно повышает ценность диагностической информации и формирует у врача клиническое мышление. Первый том посвящен УЗИ в акушерстве.
Рассматриваются ультразвуковые параметры матери и плода на разных сроках гестации, различные варианты патологии беременности.
Отдельные главы посвящены ультразвуковой допплерографии и трехмерному УЗИ в режиме реального времени, ультразвуковой поддержке инвазивных диагностических и лечебных процедур. В приложении дается подробная справочная фетометрическая информация.
Книга предназначена для акушеров-гинекологов, специалистов по ультразвуковой диагностике женских консультаций и акушерских стационаров, студентов медицинских вузов и факультетов.
Книга "Ультразвуковая диагностика в акушерстве и гинекологии: в 2 томах. Том 2. Гинекология" - Мерц Эберхард
Данное издание является наиболее полным иллюстрированным руководством по ультразвуковой диагностике в акушерстве и гинекологии. Авторы постарались охватить все известные на сегодняшний день режимы УЗИ, но тем не менее основной акцент делается на наиболее доступных методиках, технике, глубоком анализе результатов. Результаты ультразвуковых исследований приводятся в тесной корреляции с клиническими и лабораторными данными, что значительно повышает ценность диагностической информации и формирует у врача клиническое мышление.
Второй том посвящен проблемам ультразвуковых исследований в гинекологической практике: УЗ-анатомии здоровой женщины, порокам развития и заболеваниям женского полового тракта. Отдельные главы посвящены трансвагинальному и трансректальному методам исследования, а также УЗИ молочных желез.
Книга предназначена для акушеров-гинекологов, специалистов по ультразвуковой диагностике женских консультаций и акушерских стационаров, студентов медицинских вузов и факультетов.
Книга "Основы ультразвукового исследования сосудов" - В. П. Куликов
Руководство «Основы ультразвукового исследования сосудов» предназначено для тех, кто хотел бы получить по возможности краткую, но достаточно полную и главное практически полезную информацию по ультразвуковой диагностике сосудистой патологии. Автор, профессор Куликов Владимир Павлович, известен специалистам по первой в России книге, посвященной дуплексному сканированию сосудов, и руководству для врачей по ультразвуковой диагностике сосудистых заболеваний. В Руководстве представлены важнейшие сведения о технике исследования, ультразвуковых критериях нормы и патологии кровеносных сосудов, основанные на международных согласительных документах и практическом опыте работы автора. Особое внимание уделено стандартизации техники, объема и терминологии описания ультразвукового исследования сосудов. Книга предназначена для врачей ультразвуковой и функциональной диагностики, сосудистых хирургов, неврологов и кардиологов, а так же для студентов и врачей, обучающихся по программам ультразвукового исследования сосудов.
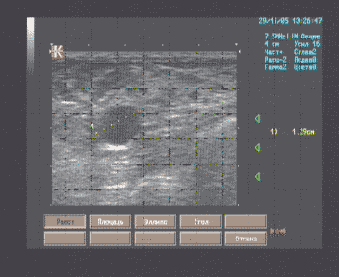
Протокол ультразвукового исследования молочных желез
(образец протокола УЗИ, ультразвукового описания патологии молочных желез)
Молочные железы симметричные, средних размеров, кожные покровы не изменены, втяжение сосков не отмечается. Позадисосковая область визуализируется в косых проекциях, типичного строения. В заднем отделе верхнего наружно квадранта левой молочной железы на 11 часах в 1 см. от соска лоцируется жидкостная полость диаметром 12 мм. с ровными контурами, не имеющая внутренних отражений, псевдоусилением сигнала задней стенки; вблизи полости отдельные млечные протоки расширены до 4-4,5 мм. На остальном протяжении дифференциация тканей молочных желез хорошая, соотношение фиброгландуллярной ткани с жировой и состояние млечных протоков соответствуют фазе менструального цикла ( диаметр млечных протоков 1 – 2,5 мм.), нарушение архитектоники не отмечается. Лимфоузлы и лимфатические протоки в регионарных зонах лимфооттока не визуализируются.
Заключение
Эхографические признаки кисты, очаговой дилатации млечных протоков левой молочной железы.
Ультразвуковая диагностика не является основным
методом и требует подтверждения
диагноза другими методами обследования.
Книга "Практическое руководство по ультразвуковой диагностике. Общая ультразвуковая диагностика" - В. В. Митьков
Фундаментальное клиническое руководство подготовлено коллективом ведущих специалистов ультразвуковой диагностики. В книге представлены разделы, посвященные ультразвуковым диагностическим системам, физическим принципам ультразвуковой диагностики, ультразвуковой диагностике заболеваний печени, желчевыводящей системы, поджелудочной железы, пищевода, желудка, кишечника, селезенки, почек, мочевого пузыря, предстательной железы и семенных пузырьков, надпочечников, органов мошонки, лимфатической системы, молочных, щитовидной, околощитовидных и слюнных желез, органов грудной клетки. Книга предназначена для врачей ультразвуковой диагностики, рентгенологов, радиологов, терапевтов, гастроэнтерологов, эндокринологов, хирургов, урологов, и всех заинтересованных специалистов.
В 3 издании монографии «Эхография в гинекологии» рассмотрены все основные вопросы ультразвуковой диагностики в гинекологии, с которыми ежедневно сталкивается врач, обследующий органы малого таза у женщин в амбулаторной практике и гинекологическом стационаре. Внесены дополнения результатов собственных научных исследований, а также опыта работы ведущих лабораторий мира и нашей страны за последнее время. Особое внимание уделено вопросам стандартизации при обследовании миометрия, эндометрия и яичников, основанных на рекомендациях групп международных экспертов. Написаны новые главы, посвященные послеродовому периоду в норме и при осложнениях, ультразвуковому мониторингу при проведении аборта как медикаментозного, так и путем вакуум-аспирации, а также послеабортным и послеоперационным осложнениям, включая проблему рубца на матке. Каждая глава состоит из небольшого этио-патогенетического раздела, подробно освещены вопросы эхографической диагностики, включая данные цветового картирования, допплерометрии, новых, недостаточно распространённых методик и дифференциально-диагностические критерии. Каждая глава иллюстрирована большим количеством эхограмм как типичного, так и нетипичного изображения рассматриваемой патологии. Определены диагностические возможности эхографии, цветового картирования и допплерометрии во всех рассматриваемых разделах гинекологии. Представлены новые направления диагностики и лечения, внедряемые в гинекологическую практику в течение последних лет. В приложение включены таблицы всех нормативных параметров, предложены протоколы ультразвукового исследования органов малого таза и проведения эхогистеросальпингоскопии. Книга рассчитана на врачей ультразвуковой диагностики, гинекологов, акушеров, онкогинекологов, хирургов и врачей смежных специальностей.
Книга "Эхокардиография от Рыбаковой" - М. К. Рыбакова, В. В. Митьков
Данное издание представляет собой практическое руководство по эхокардиографии, в котором отражены все современные технологии, применяемые в настоящее время. Исключительный интерес для специалистов представляет CD-ROM с подборкой видеоклипов по всем основным разделам, включающих редкие случаи диагностики.
Особенность издания - попытка объединить и сравнить результаты эхокардиографического исследования сердца и паталогоанатомический материал по всем основным разделам. Большой интерес представляют разделы, содержащие новые технологии исследования, такие как трех- и четырехмерная реконструкция сердца в реальном времени, тканевая допплерография. Большое внимание уделено также классическим разделам эхокардиографии – оценке легочной гипертензии, клапанных пороков сердца, ишемической болезни сердца и ее осложнений и т.д.
В книге представлены огромный иллюстративный материал, большое количество схем и рисунков, приведены алгоритмы тактики проведения исследования и диагностики по всем разделам эхоКГ. Руководство помогает разрешить спорные и злободневные вопросы, позволяет ориентироваться в расчетах и измерениях, содержит необходимую справочную информацию. Книга написана сотрудниками кафедры ультразвуковой диагностики ГБОУ ДПО «Российская медицинская академия последипломного образования'' Министерства здравоохранения Российской Федерации» (база – ГКБ им. С.П. Боткина, Москва).
Издание предназначено для специалистов эхокардиографии, врачей ультразвуковой и функциональной диагностики, кардиологов и терапевтов.
Книга "УЗИ в акушерстве и гинекологии. Том 1 Акушерство" - Мерц Эберхард
Данное издание является наиболее полным иллюстрированным руководством по ультразвуковой диагностике в акушерстве и гинекологии.
Авторы постарались охватить все известные на сегодняшний день режимы УЗИ, но тем не менее основной акцент делается на наиболее доступных методиках, технике, глубоком анализе результатов. Результаты ультразвуковых исследований приводятся в тесной корреляции с клиническими и лабораторными данными, что значительно повышает ценность диагностической информации и формирует у врача клиническое мышление. Первый том посвящен УЗИ в акушерстве.
Рассматриваются ультразвуковые параметры матери и плода на разных сроках гестации, различные варианты патологии беременности.
Отдельные главы посвящены ультразвуковой допплерографии и трехмерному УЗИ в режиме реального времени, ультразвуковой поддержке инвазивных диагностических и лечебных процедур. В приложении дается подробная справочная фетометрическая информация.
Книга предназначена для акушеров-гинекологов, специалистов по ультразвуковой диагностике женских консультаций и акушерских стационаров, студентов медицинских вузов и факультетов.
Книга "Ультразвуковая диагностика в акушерстве и гинекологии: в 2 томах. Том 2. Гинекология" - Мерц Эберхард
Данное издание является наиболее полным иллюстрированным руководством по ультразвуковой диагностике в акушерстве и гинекологии. Авторы постарались охватить все известные на сегодняшний день режимы УЗИ, но тем не менее основной акцент делается на наиболее доступных методиках, технике, глубоком анализе результатов. Результаты ультразвуковых исследований приводятся в тесной корреляции с клиническими и лабораторными данными, что значительно повышает ценность диагностической информации и формирует у врача клиническое мышление.
Второй том посвящен проблемам ультразвуковых исследований в гинекологической практике: УЗ-анатомии здоровой женщины, порокам развития и заболеваниям женского полового тракта. Отдельные главы посвящены трансвагинальному и трансректальному методам исследования, а также УЗИ молочных желез.
Книга предназначена для акушеров-гинекологов, специалистов по ультразвуковой диагностике женских консультаций и акушерских стационаров, студентов медицинских вузов и факультетов.
Книга "Основы ультразвукового исследования сосудов" - В. П. Куликов
Руководство «Основы ультразвукового исследования сосудов» предназначено для тех, кто хотел бы получить по возможности краткую, но достаточно полную и главное практически полезную информацию по ультразвуковой диагностике сосудистой патологии. Автор, профессор Куликов Владимир Павлович, известен специалистам по первой в России книге, посвященной дуплексному сканированию сосудов, и руководству для врачей по ультразвуковой диагностике сосудистых заболеваний. В Руководстве представлены важнейшие сведения о технике исследования, ультразвуковых критериях нормы и патологии кровеносных сосудов, основанные на международных согласительных документах и практическом опыте работы автора. Особое внимание уделено стандартизации техники, объема и терминологии описания ультразвукового исследования сосудов. Книга предназначена для врачей ультразвуковой и функциональной диагностики, сосудистых хирургов, неврологов и кардиологов, а так же для студентов и врачей, обучающихся по программам ультразвукового исследования сосудов.

")


 прободает околоушную железу (GP) и служит ключевым внутренним ориентиром для поиска шейной")
 располагается сверху и кпереди от позадинижнечелюстной")

")
")

 с его сухожилием (T-MFCU) идут поверх тыльной ветви сразу после ее отхождения от локтевого нерва")

")


 отбрасывает характерную дорсальную тень")
 визуализируется в области ТОВ (между сосудами и превертебральными мышцами)")


 и нити псевдомицелия рода Candida в мазке из материала от больного кандидозом")


 регуляции женской репродуктивной системы. ГнИГ- гонадотропинингибирующий гормон")